Congratulations to Robert Kelly, Frank Schnorrer, Cédric Maurange, Bianca Habermann and Delphine Delacour!

IBDM Marseille inspires young minds: engaging primary school children on childhood cancer (“Contre le cancer, j’apporte ma pierre”) and interacting with high school students through immersive experiences (DECLICS).

Join us on 29/06/2023 at 12:30 in Amphi 12 for an exciting talk by Rikesh Jain and Theo Brunet from our Team!
5 motivated and talented students successfully defended their thesis between September 2022 and January 2023.

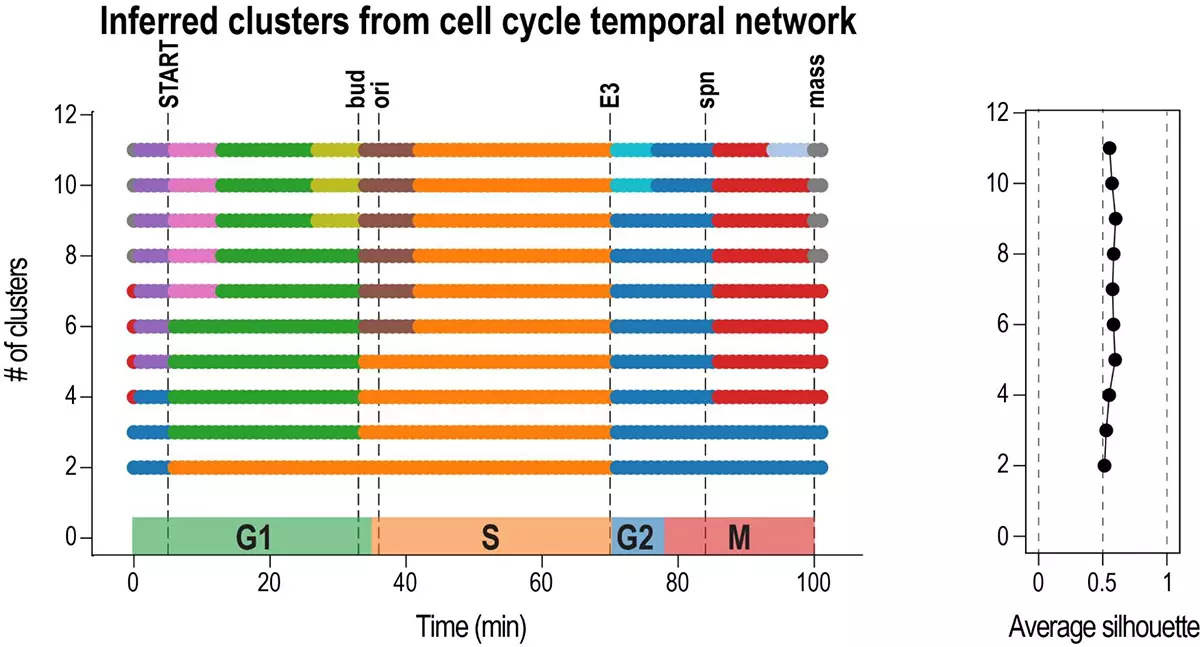
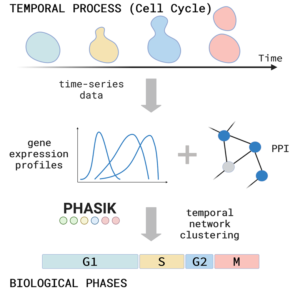
Show me your rhythm!
We introduce an algorithm, Phasik, for extracting the phases of biological systems by clustering partial temporal networks.
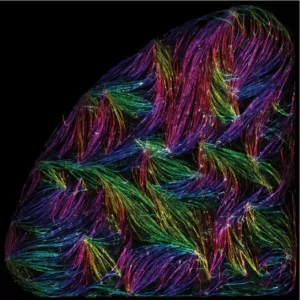
Self-organisation of human muscles in a dish
Human muscle cells self-organise into defined fiber bundles in vitro even without the presence of external cues !
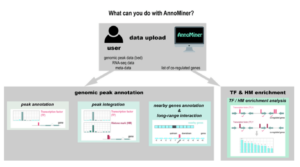
We introduce a novel, user-friendly web-based tool ‘AnnoMiner’ to annotate and integrate epigenetic and transcription factor binding data.
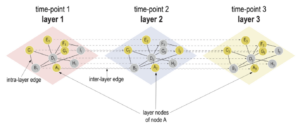
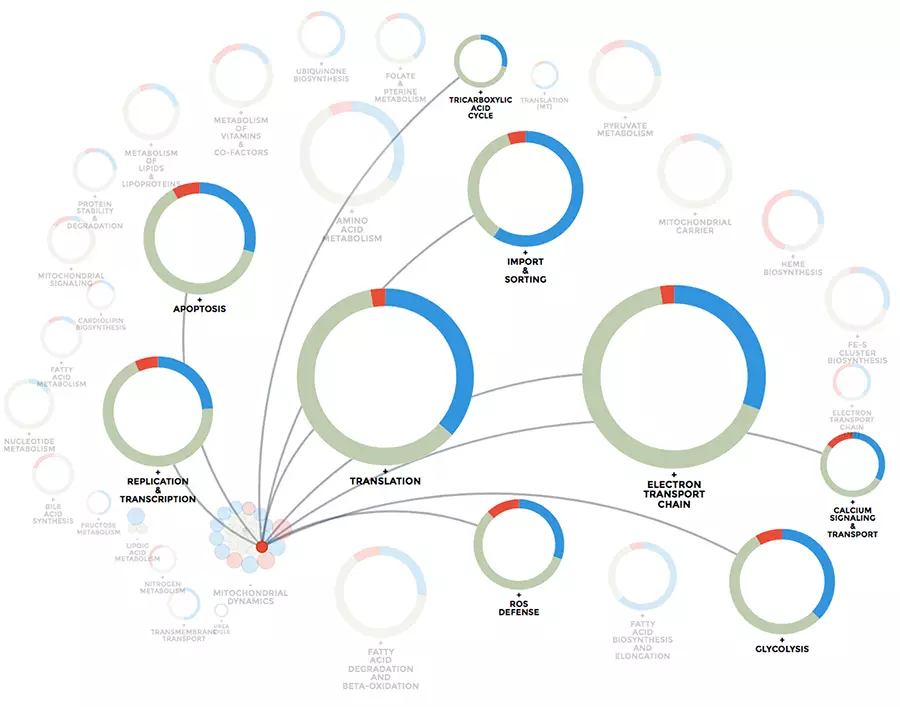
Look at the TIME in your interaction network
The Habermann team has repurposed the concept of multilayer networks generally used to integrate different types of data.