Félicitation à Robert Kelly, Frank Schnorrer, Cédric Maurange, Bianca Habermann et Delphine Delacour !

L’IBDM inspire les jeunes esprits : en impliquant les enfants des écoles primaires dans la lutte contre le cancer pédiatrique (“Contre le cancer, j’apporte ma pierre”) et en interagissant avec les lycéens grâce à des expériences immersives (DECLICS).

Rejoignez-nous le 29/06/2023 à 12:30 dans l’Amphi 12 pour une présentation passionnante deux des membres de notre équipe : Rikesh Jain et Theo Brunet !
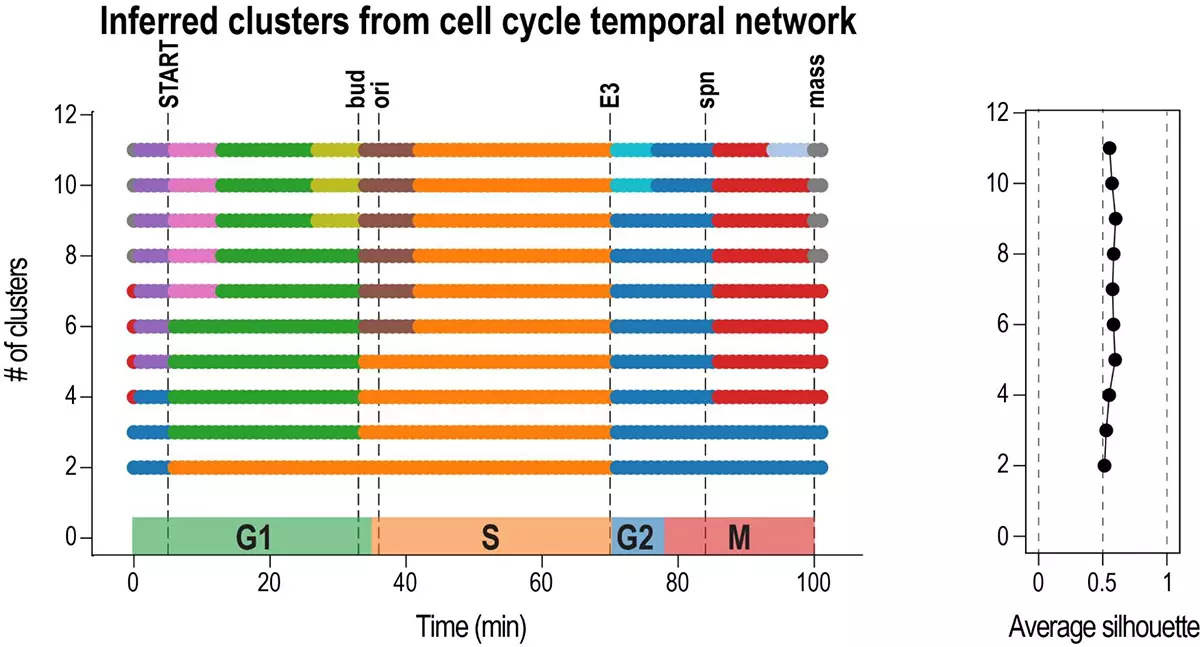
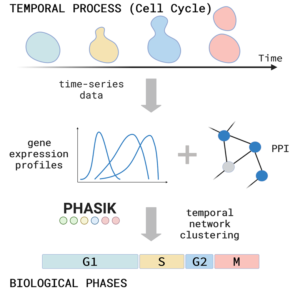
Montre-moi ton rythme !
Phasik est un algorithme permettant d’extraire les phases des systèmes biologiques en regroupant des réseaux temporels partiels.
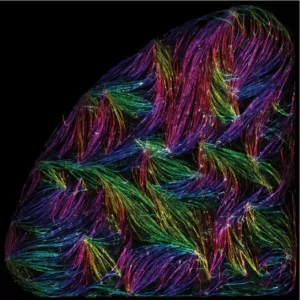
Les cellules musculaires s’auto-organisent en faisceaux de fibres in vitro, sans la présence de signaux externes !
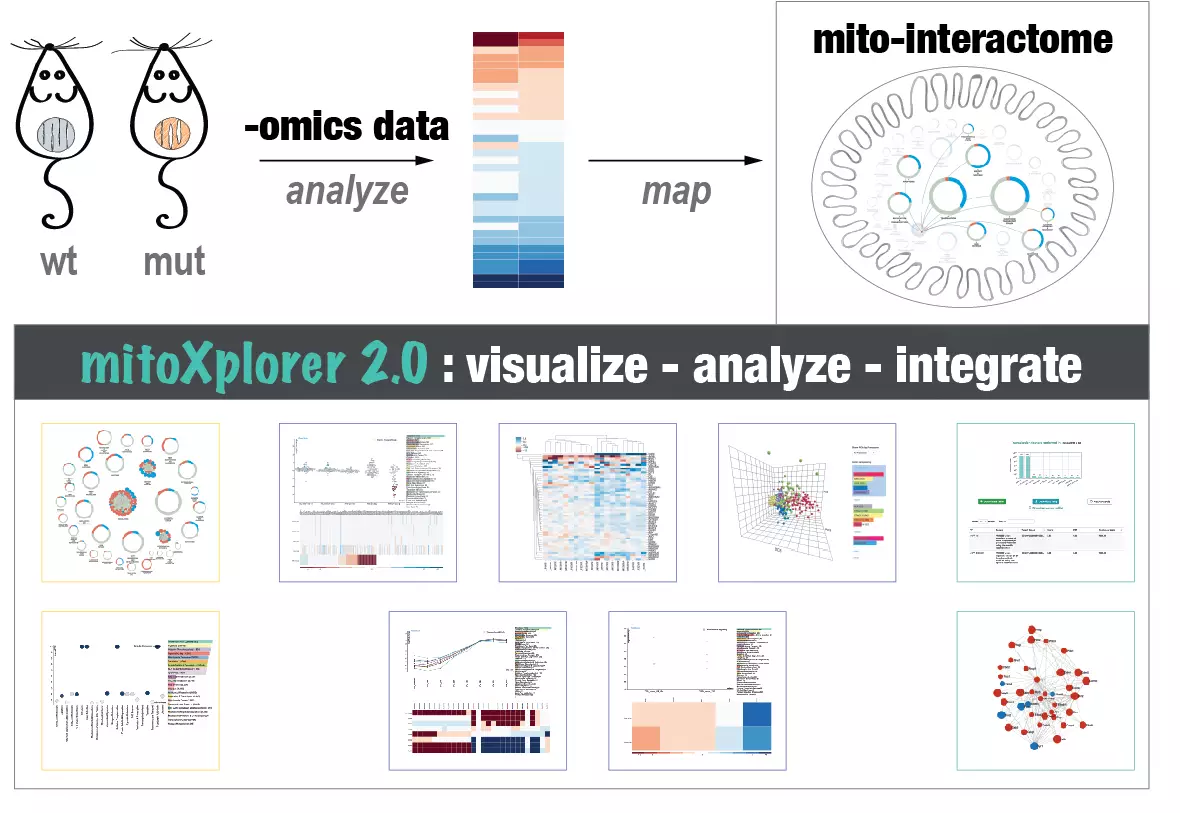
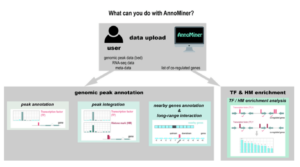
Nous présentons ‘AnnoMiner’, un nouvel outil convivial basé sur le web pour annoter et intégrer les données épigénétiques et de liaison des facteurs de transcription.
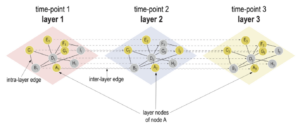
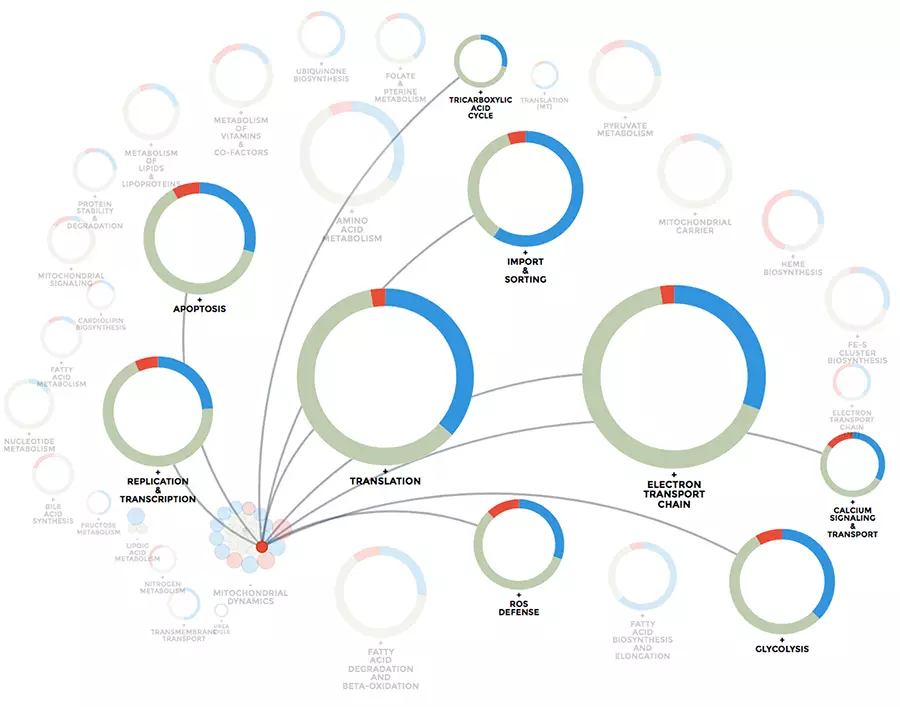
L’équipe Habermann a adapté le concept des réseaux multicouches généralement utilisés pour intégrer différents types de données.